Our Research
Discovery of New Therapeutic Targets in Leukemia and Other Blood Cancers

One of our major goals is to discover innovative therapeutic strategies for aggressive blood cancers, such as acute myeloid leukemia (AML), via improved understanding of disease biology, determinants of response to therapy, and mechanisms of treatment resistance. In a recent study (publication link: Nature), we used functional genomics and CRISPR interference screening to uncover a targetable dependency on the PI3Kγ pathway across acute leukemias. This dependency was enriched in high-risk AMLs with monocytic and dendritic differentiation and in cases with high expression of the PI3Kγ adapter protein, PIK3R5. Surprisingly, survival signaling downstream of PI3Kγ in these dependent leukemias was via activation of the kinase PAK1 rather than AKT. We also found that PIK3R5-high leukemias were sensitive to treatment in vivo with the PI3Kγ-selective drug eganelisib, and both PIK3R5 high and low leukemias showed additive or synergistic efficacy in vivo when eganelisib was combined with the leukemia chemotherapy cytarabine. We continue to refine our understanding of mechanisms of sensitivity and resistance to inhibition of PI3Kγ and we are advancing eganelisib into clinical trials for patients with AML.
In another recent project, we explored mechanisms to enhance the adaptive immune response to AML (Blood Advances). We investigated the role of AMPK, 5' adenosine monophosphate-activated protein kinase, a central regulator of cellular energy homeostasis, in inducing an immunogenic form of cell death. We found that pharmacologic activation of AMPK promotes cell surface exposure of calreticulin, a key 'eat-me' signal for immune cells, and in doing so enhances the immunogenicity of AML cells.
In another recent project, we explored mechanisms to enhance the adaptive immune response to AML (Blood Advances). We investigated the role of AMPK, 5' adenosine monophosphate-activated protein kinase, a central regulator of cellular energy homeostasis, in inducing an immunogenic form of cell death. We found that pharmacologic activation of AMPK promotes cell surface exposure of calreticulin, a key 'eat-me' signal for immune cells, and in doing so enhances the immunogenicity of AML cells.
Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN)
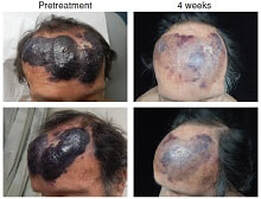
BPDCN is a highly aggressive hematologic malignancy of plasmacytoid dendritic cells, related to AML, which exhibits both skin and bone marrow involvement. BPDCN has no standard therapy and is nearly uniformly fatal within two years from diagnosis (publication link: Blood). The genetic and epigenetic alterations driving BPDCN are not well understood.
We are studying the genomics of BPDCN and modeling how specific somatic alterations, including RNA splicing factor and heterotrimeric G protein mutations, contribute to dendritic cell transformation. We discovered that BPDCN is highly and uniquely dependent on the antiapoptotic protein BCL2 for survival, and that BPDCN is markedly sensitive to BCL2 inhibition with venetoclax (Cancer Discovery). We are leading clinical trials of venetoclax in BPDCN and testing patient samples from those trials in the laboratory.
We have performed phylogenomics with whole genome DNA sequencing coupled with single cell RNA seq/single cell genotyping to discover ultraviolet radiation in TET2 mutated clonal hematopoiesis cells as a path to development of BPDCN (Nature). We also used single cell dendritic cell and T cell receptor immunoprofiling, as well as microbial metagenomics to understand BPDCN biology (Science, Frontiers in Immunology, Blood Advances). We are also conducting clinical trials in patients with BPDCN and AML; we led a multicenter trial testing the CD123/IL3 receptor-targeting immunotoxin tagraxofusp that led to the first ever FDA approval for patients with BPDCN (New England Journal of Medicine). We determined how AMLs and BPDCNs become resistant to tagraxofusp via epigenetic downregulation of the diphthamide synthesis pathway (Journal of Clinical Investigation). This led to an ongoing clinical trial combining tagraxofusp with azacitidine and venetoclax in AML, MDS, and BPDCN.
We are studying the genomics of BPDCN and modeling how specific somatic alterations, including RNA splicing factor and heterotrimeric G protein mutations, contribute to dendritic cell transformation. We discovered that BPDCN is highly and uniquely dependent on the antiapoptotic protein BCL2 for survival, and that BPDCN is markedly sensitive to BCL2 inhibition with venetoclax (Cancer Discovery). We are leading clinical trials of venetoclax in BPDCN and testing patient samples from those trials in the laboratory.
We have performed phylogenomics with whole genome DNA sequencing coupled with single cell RNA seq/single cell genotyping to discover ultraviolet radiation in TET2 mutated clonal hematopoiesis cells as a path to development of BPDCN (Nature). We also used single cell dendritic cell and T cell receptor immunoprofiling, as well as microbial metagenomics to understand BPDCN biology (Science, Frontiers in Immunology, Blood Advances). We are also conducting clinical trials in patients with BPDCN and AML; we led a multicenter trial testing the CD123/IL3 receptor-targeting immunotoxin tagraxofusp that led to the first ever FDA approval for patients with BPDCN (New England Journal of Medicine). We determined how AMLs and BPDCNs become resistant to tagraxofusp via epigenetic downregulation of the diphthamide synthesis pathway (Journal of Clinical Investigation). This led to an ongoing clinical trial combining tagraxofusp with azacitidine and venetoclax in AML, MDS, and BPDCN.
Sex Bias In Cancer
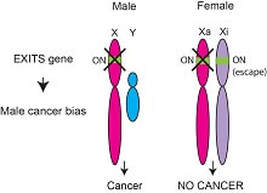
150,000 more men get cancer yearly in the US than women. Most individual cancer types have a male bias, including leukemia and some like BPDCN, kidney cancer, and bladder cancer that are >2:1 M:F biased. We recently examined male bias in cancer mutations across >4100 patient’s tumors and discovered a new type of tumor suppressor gene that we called Escape from X-Inactivation Tumor Suppressor (EXITS) genes (publication link: Nature Genetics). Female cells express two copies of these genes, whereas males only express one. Therefore, female cells are relatively protected from cancer because they require two mutations to inactivate these genes, while males only need one. We have searched for novel sex-associated cancer genes and characterized the function of ZRSR2 mutations with extreme male bias in myeloid malignancies (Cancer Discovery), studied whole genomes from a leukemia of eosinophils with >9:1 male bias (Blood Advances), and investigated the role of loss of the Y chromosome in primary tumors from male patients (Cell).
Mechanisms of Clonal Dominance and Disordered Gene Regulation In Leukemia

Despite remarkable recent progress in cataloging the genomic changes associated with hematologic malignancies, we do not understand how many “driver” mutations contribute to disease development nor how they combine with other alterations to result in leukemia. We have implicated the nucleosome regulator HMGN1, a target of recurrent amplification in leukemia, as a myeloid and lymphoid leukemia oncogene that causes widespread epigenetic alterations including global changes in histone H3 lysine 27 acetylation and methylation at stem cell genes (publication links: Nature Genetics, Nature Communications, Cell Reports).
Leukemia-associated changes often confer a clonal advantage to hematopoietic stem and/or progenitor cells. We are developing in vivo model systems to understand how these changes gain a selective advantage, and we are using those platforms to develop therapies to target clonal dominance that may avert or delay progression to cancer. As one example, we discovered and determined the mechanism of transformation by GNB1 and GNB2 mutations seen in AML, MDS, BPDCN, and lymphomas (Nature Medicine), and are working to understand other mechanisms that promote clonal outgrowth and subsequent malignancy.
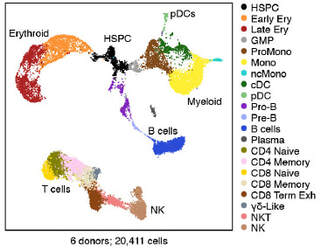
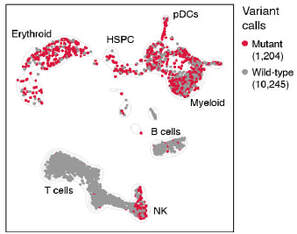
We also use single cell RNA-sequencing with multi-omics to profile leukemia at individual cell resolution (Nature), including using a technique we developed called XV-seq (eXpressed Variant sequencing). We are exploiting these technologies and developing others for capturing and interrogating single ultra-rare cells in the setting of measurable/minimal residual disease (MRD) after therapy for AML and lymphoma.
Leukemia-associated changes often confer a clonal advantage to hematopoietic stem and/or progenitor cells. We are developing in vivo model systems to understand how these changes gain a selective advantage, and we are using those platforms to develop therapies to target clonal dominance that may avert or delay progression to cancer. As one example, we discovered and determined the mechanism of transformation by GNB1 and GNB2 mutations seen in AML, MDS, BPDCN, and lymphomas (Nature Medicine), and are working to understand other mechanisms that promote clonal outgrowth and subsequent malignancy.
We also use single cell RNA-sequencing with multi-omics to profile leukemia at individual cell resolution (Nature), including using a technique we developed called XV-seq (eXpressed Variant sequencing). We are exploiting these technologies and developing others for capturing and interrogating single ultra-rare cells in the setting of measurable/minimal residual disease (MRD) after therapy for AML and lymphoma.
|
|